近日,美国食品药品监督管理局(FDA)正式批准UCB公司研发的 KYGEVVI 上市,用于治疗症状起始年龄不超过12岁的胸苷激酶2缺乏症(TK2d) 患者。这是全球首个、也是唯一一个获得批准的TK2d针对性疗法,为这一罕见且致命的线粒体疾病带来了真正意义上的治疗希望。
罕见而致命的TK2缺乏症:能量枯竭的悲剧
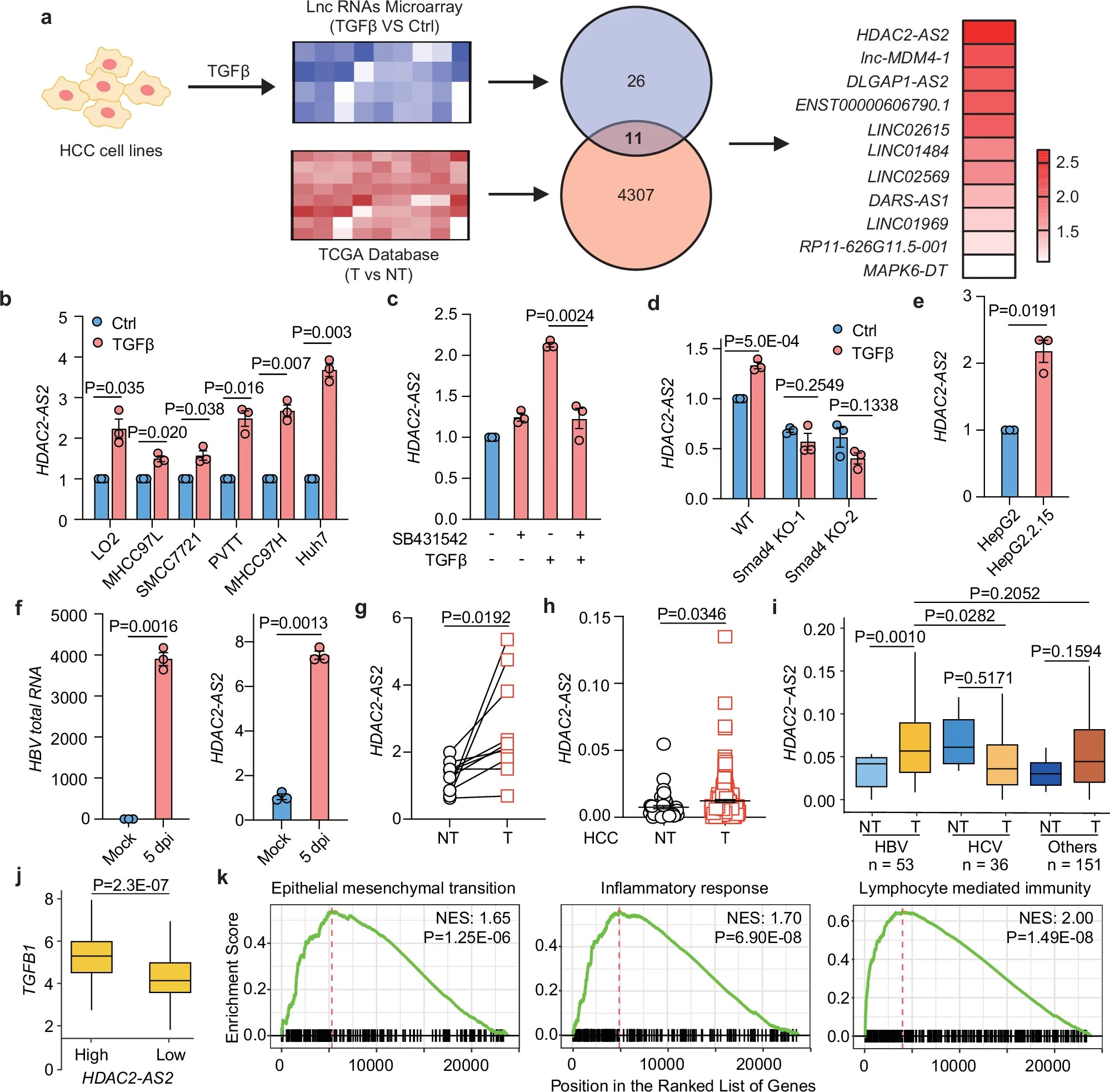
胸苷激酶2缺乏症(TK2d)是一种极为罕见的线粒体DNA合成障碍疾病,全球发病率约为每百万人1.64例。患者多在婴幼儿时期发病,表现为肌无力、吞咽困难、呼吸衰竭等症状。随着病情进展,患者会逐渐丧失行走、吞咽甚至自主呼吸能力。尤其是发病年龄在12岁以前的儿童,死亡风险极高,大多数患者在确诊后3年内不幸离世。
长期以来,医学界对TK2d束手无策,除支持治疗外无任何获批药物,患者及家庭长期面临生理与心理的双重煎熬。
发病根源:线粒体DNA“燃料”中断
要理解TK2d的本质,就需要从细胞的能量中枢——线粒体 说起。线粒体负责为细胞提供能量,而DNA在其中的复制和修复过程离不开一种关键酶——胸苷激酶2(TK2)。
TK2的主要任务是催化嘧啶核苷酸的磷酸化反应,为线粒体DNA合成提供原料。当TK2基因发生突变后,酶活性下降或完全丧失,导致线粒体DNA的原料供应被切断。
这就像一座城市的“电力站”缺乏燃料——细胞失去能量供应,尤其是能量需求极高的肌肉细胞首当其冲,最终出现严重的能量代谢障碍,引发肌无力、呼吸衰竭等一系列致命症状。

KYGEVVI的创新突破:开辟“代谢旁路”,让能量恢复流动
KYGEVVI的独特之处在于采用了代谢旁路疗法(metabolic bypass therapy)。该药物由两种核苷类似物——doxecitine 和 doxribtimine 组成,它们能绕过失效的TK2酶,直接补充线粒体DNA合成所需的底物(脱氧胞苷和脱氧胸苷)。
换句话说,KYGEVVI在“堵塞”的代谢通路旁修建了一条新的能量补给通道,使线粒体仍能获得所需原料并继续合成DNA,从根源上恢复线粒体的功能。
这项疗法的出现,不仅能显著改善患者的肌肉功能与呼吸能力,更被视为线粒体遗传病治疗史上的里程碑式突破。
从支持到治本:TK2d治疗的重大转折
以往TK2d患者只能依靠呼吸辅助、营养支持等方式延缓病程。而KYGEVVI的问世,则意味着治疗策略正式从“缓解症状”跨越到“修复病因”。
这不仅是罕见病治疗领域的一次重要胜利,也为未来其他线粒体疾病的精准治疗提供了新方向。UCB公司在新闻稿中表示,KYGEVVI的获批为罕见遗传代谢病患者带来了新的生命希望,标志着“代谢旁路疗法”正式进入临床应用阶段。
结语:为罕见病患者点亮希望之光
TK2缺乏症曾是医学界几乎无药可治的“黑洞”,如今,KYGEVVI的成功上市不仅填补了这一领域的空白,更象征着罕见病精准治疗的新时代已经到来。
这款创新药的诞生告诉我们:即使面对最稀有、最复杂的遗传疾病,科学也能找到通往希望的“旁路”。未来,随着更多创新疗法的出现,或许“罕见病”将不再意味着“无药可医”。