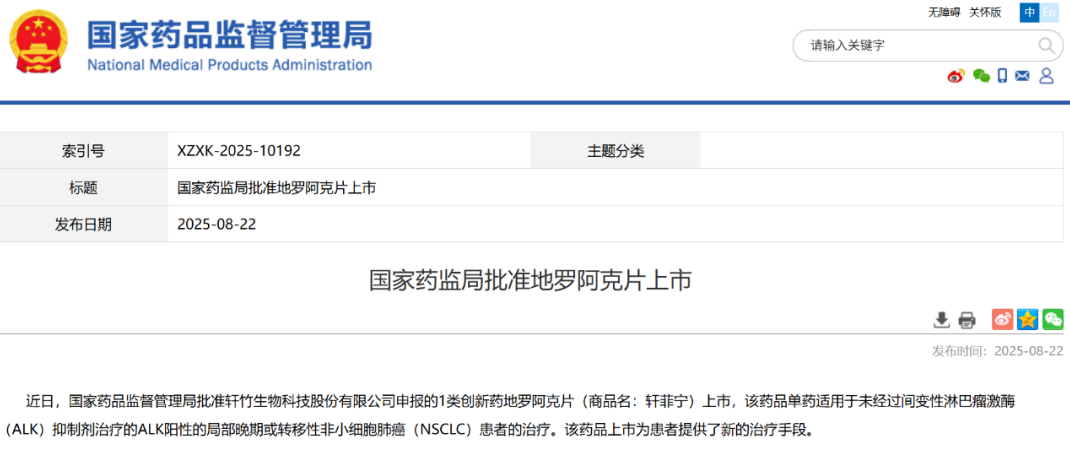
2025年8月19日,深圳普瑞金生物药业股份有限公司(简称“普瑞金”)自主研发的 靶向CD19&CD22的CAR-T注射液
荣获美国食品药品监督管理局(FDA)授予的 孤儿药资格认定(Orphan Drug Designation, ODD),用于治疗
急性淋巴细胞白血病(ALL)。这一资质认可标志着普瑞金在CAR-T抗肿瘤疗法领域的巨大潜力,同时为难治性血液恶性肿瘤患者带来了新的希望。 CAR-T双靶点创新:提高疗效与减少耐药风险 普瑞金开发的靶向CD19&CD22
CAR-T注射液是一款处于临床前的细胞治疗创新产品,具备完全自主知识产权。目前正在与国内三甲医院血液科团队合作开展
患者主导试验(IIT),初步数据令人振奋。 临床数据: 完全缓解(CR):针对首批接受该疗法治疗的6名患者,均获得了完全缓解,治疗效果显著。 客观缓解率(ORR):达到 100%,所有患者病情均得以明显改善。 微小残留病(MRD)阴性率:成功实现 100% 阴性率,表明治疗能有效清除癌细胞。 这种创新型CAR-T疗法通过联合靶向
CD19和CD22,解决了单一靶点治疗的潜在耐药问题,为复发或难治性急性淋巴细胞白血病提供了一种多靶点的治疗选择。 急性淋巴细胞白血病(ALL)治疗现状:亟需突破性疗法 急性淋巴细胞白血病(ALL)是一种源于淋巴细胞的血液系统恶性肿瘤,其发生机制与基因突变阻碍正常分化和凋亡有关,导致癌细胞无限增殖。 ALL分类与特点: B-ALL:占比超过 80% 的ALL病例,发生于B细胞祖细胞阶段,是儿童和成人急性白血病的重要类型。 T-ALL:起源于T细胞前体,发病比例相对较低。 当前治疗困境: 传统疗法有限:化疗及靶向药物虽能缓解部分患者病情,但复发及难治性B-ALL患者预后极差。 复发/难治性患者生存期短:现有治疗手段难以根本改善这部分患者的生存率,迫切需要新的治疗策略。 靶向CD19&CD22 CAR-T疗法的临床与应用价值 普瑞金的双靶点CAR-T治疗策略,通过同时靶向 CD19和CD22,克服了单靶点治疗耐药性高及疗效局限等问题: 双靶点联合优势: CD19和CD22均为B细胞急性淋巴细胞白血病的特异性标志物,通过联合靶向有效提高治疗覆盖率。 减少癌细胞耐药机制,克服复发/难治性B-ALL的治疗瓶颈。 孤儿药认证助力: FDA授予孤儿药资格(ODD),强化了该疗法在治疗稀缺难治性疾病中的临床应用前景。 孤儿药认证不仅为普瑞金提供了研发支持,还将加速其国际拓展步伐。 市场前景与临床需求 复发/难治性B-ALL患者的亟需选择: 复发或难治性B-ALL患者长期面临有限治疗选择,而普瑞金的靶向CD19&CD22
CAR-T注射液通过显著性疗效及安全性数据,表现出了令人期待的突破性表现。 全球市场潜力: 急性淋巴细胞白血病作为一种儿童及成人均高发的恶性血液肿瘤,具有广阔的治疗市场。CAR-T疗法近年来在抗肿瘤领域持续快速增长,普瑞金的双靶点疗法有望结合作为新标准进入国际市场。 总结:开启ALL治疗新篇章 普瑞金开发的靶向CD19&CD22
CAR-T注射液通过有力的临床数据及FDA孤儿药资格认证,正展现出显著的治疗潜力,尤其为复发/难治性急性淋巴细胞白血病患者提供了新的治疗选择。 未来,该疗法的进一步临床试验及大规模研发推广,有望显著改善ALL患者生存质量,同时成为CAR-T抗肿瘤疗法领域的国产创新典范,为全球AML治疗带来更多可能性。