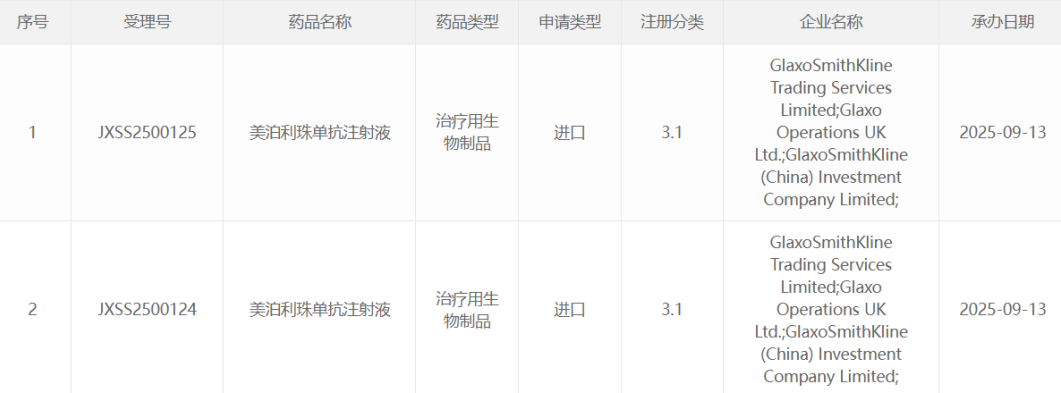
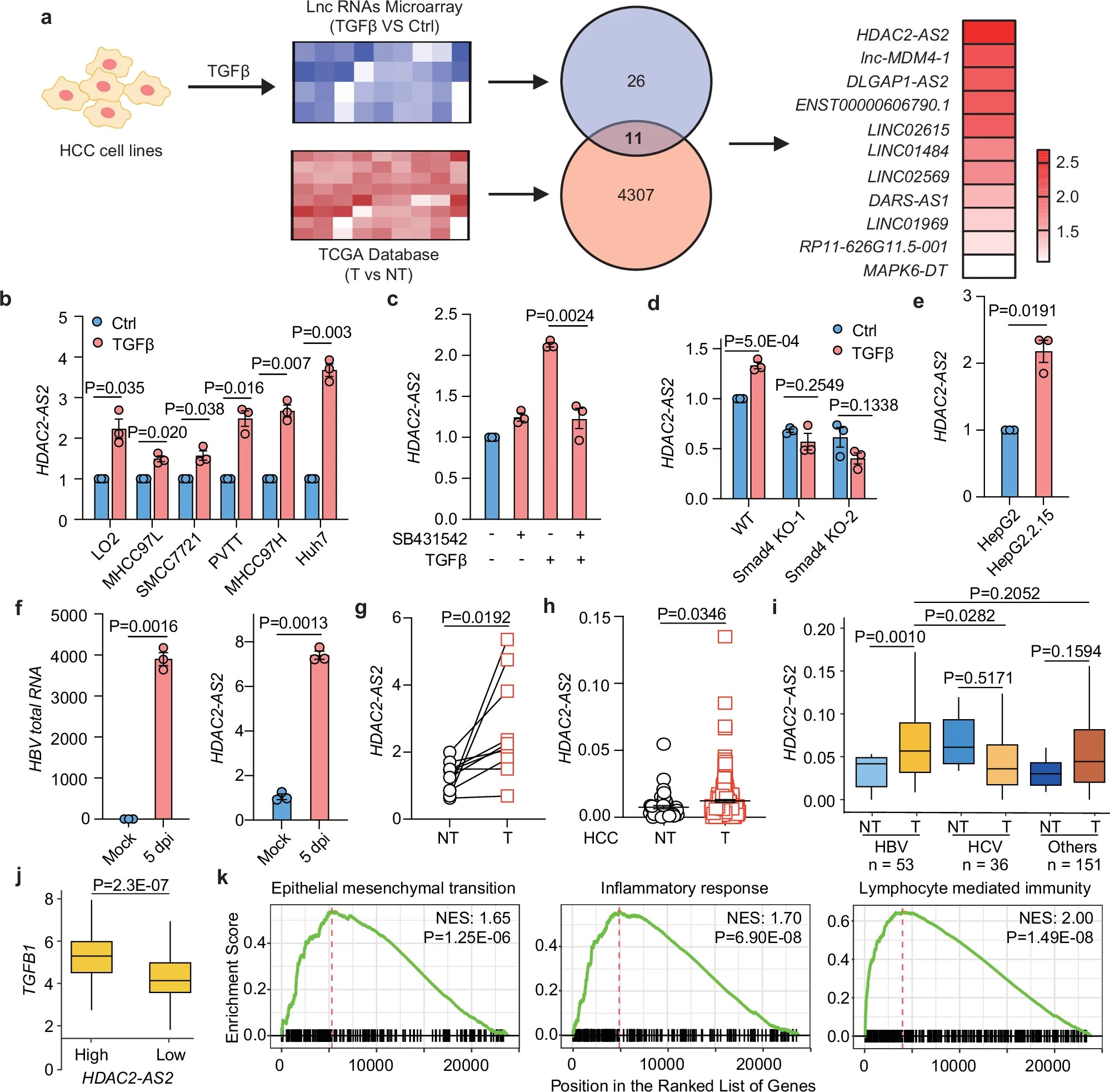
对于很多家长而言,孩子持续癫痫发作、语言和运动发育迟缓却找不到病因,这无疑是一种巨大的心理负担。这类症状通常和神经发育障碍有关,约有20%到30%的病例可能和遗传因素相关,但很多时候病因基因难以定位。
近期,权威期刊《Brain》刊登了一项重要研究——《SSPOP基因复合杂合突变导致癫痫及发育障碍》,为这些家庭带来了新的曙光。研究首次揭示长期被认为是“无功能假基因”的SSPOP其实具有重要生物学功能,其复合杂合突变直接诱发儿童癫痫及神经发育障碍,这一结论也获得了多种实验模型的验证。

SSPOP基因不再是“假基因”,其重要功能首次被确认
长期以来,SSPOP基因因功能模糊,曾被划为“假基因”。实际上,SSPOP编码的SCO-spondin蛋白,在胚胎发育阶段对脊髓构建及神经细胞保护发挥着重要作用。它能调节脑脊液中的多个关键分子,促进神经细胞生长和抗损伤能力。然而,之前未有人明确证明SSPOP与人类疾病的直接关联,直到这项研究取得突破。
临床发现:3个无关家庭4名患儿均携带复合杂合突变
研究团队通过临床病例分析,重点关注了3个毫无血缘关系的家庭,涉及4名孩子(包括一对异卵双胞胎)。这些孩子均无明显遗传病史或出生缺陷,但均出现早期癫痫发作(最早于出生4天内发病),症状包括局部抽搐、全身僵硬及癫痫性痉挛。此外,患儿在语言及社交能力发育上明显落后,尽管脑部MRI未显示结构异常。
基因检测揭露了关键线索:所有孩子均携带SSPOP基因的复合杂合突变,即同时存在错义突变与剪接位点突变或片段缺失。错义突变扰乱了SCO-spondin蛋白结构,使其功能受损;剪接突变导致关键外显子缺失,破坏蛋白的正常生成。
多重实验证实SSPOP基因在脑发育中的核心作用
为了澄清SSPOP基因功能,团队查询BrainSpan数据库,发现该基因在出生后大脑存在高表达。通过qRT-PCR检测患儿脑组织,确认其mRNA序列与参考序列完全匹配。免疫荧光观察显示,SCO-spondin蛋白在不同年龄段儿童脑组织及体外培养的脑类器官中均存在。Western blot进一步验证,该蛋白在新生儿脑组织中分子量跨度较大,反映其分子结构的多样性与复杂性,确证SSPOP作为功能基因在人脑发育中占据重要地位。
斑马鱼模型揭示SSPOP基因缺失引发严重神经障碍及癫痫样放电
作为功能验证,研究者利用斑马鱼作为动物模型。斑马鱼的sspo基因与人类SSPOP基因高度同源。敲除该基因的斑马鱼后,86.8%出现明显畸形(如脊柱侧弯及脑发育异常),死亡率显著提高。详细观察显示,其眼间距增大,脑区体积明显缩小,同时中枢神经系统的荧光标记减少。最关键的是电生理检测发现,敲除组斑马鱼脑内出现癫痫样放电,发作频率高达每15分钟1至3次,而对照组无类似异常,强有力证明了该基因缺失与癫痫发作直接关联。
结语:SSPOP基因研究开启儿童神经疾病精准诊断和治疗新篇章
这项研究首次将SSPOP基因复合杂合突变与儿童癫痫及神经发育障碍紧密联系起来,并彻底颠覆了其“假基因”的旧说法。通过斑马鱼和脑类器官等多重模型,科学家们清晰描绘了其致病机理。对无数苦苦寻医无果的家庭来说,这一发现无疑提供了全新的诊断思路,未来SSPOP基因将可能成为临床遗传筛查的重要目标,助力更早确诊及个性化治疗方案的制定。同时,这也为发育性神经疾病的治疗靶点研究打开了新的窗口,期待进一步研究能够为患儿带来突破性疗效,改善他们的生活质量。